

Abstract
Background:
In a large pedigree with an unusual phenotype of spastic paraplegia or dystonia and autosomal dominant inheritance, linkage analysis previously mapped the disease to chromosome 2q24-2q31.
Objective:
To identify the genetic etiology and molecular basis of an unusual autosomal dominant spastic paraplegia and dystonia.
Methods:
Whole exome sequencing following linkage analysis was used to identify the genetic etiology in a large family. Cosegregation analysis was also performed. An additional 384 individuals with spastic paraplegia or dystonia were screened for pathogenic sequence variants in the adenosine triphosphate (ATP) synthase membrane subunit C locus 3 gene (ATP5MC3). The identified variant was submitted to the “GeneMatcher” program for recruitment of additional subjects. Mitochondrial functions were analyzed in patient-derived fibroblast cell lines. Transgenic Drosophila carrying mutants were studied for movement behavior and mitochondrial function.
Results:
Exome analysis revealed a variant (c.318C>G (p.Asn106Lys) (NM_001689.4) in ATP5MC3 in a large family with autosomal dominant spastic paraplegia and dystonia that cosegregated with affected individuals. No variants were identified in an additional 384 individuals with spastic paraplegia or dystonia. GeneMatcher identified an individual with the same genetic change, acquired de novo, who manifested upper limb dystonia. Patient fibroblast studies showed impaired complex V activity, ATP generation, and oxygen consumption. Drosophila carrying orthologous mutations also exhibited impaired mitochondrial function and displayed reduced mobility.
Conclusions:
A unique form of familial spastic paraplegia and dystonia is associated with a heterozygous ATP5MC3 variant that also reduces mitochondrial complex V activity.
TOC Graphic summary
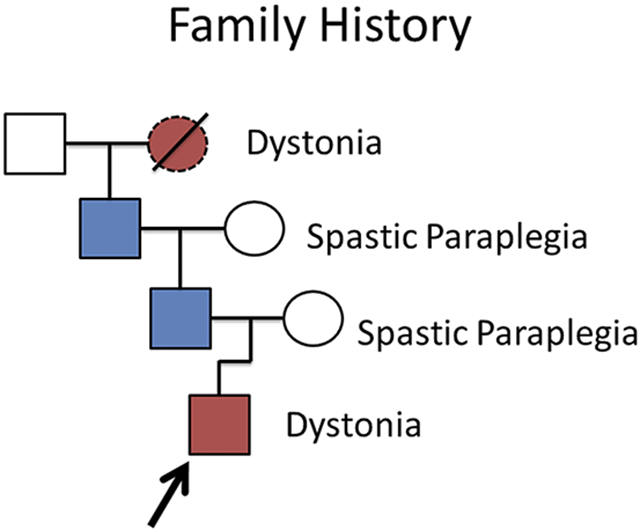
Family-1 (partial) pedigree: symptomatic proband, father, and grandfather presented for neurological consultation. Subsequent investigations identified, in two families, a variant in ATP5MC3, which encodes a component of complex V in the mitochondrial electron transport chain. Functional studies in patient-derived fibroblasts and drosophila support a loss of function.
Graphical Abstract
Introduction
Dystonia and spastic paraplegia denote neurological conditions with distinct signs and neuroanatomic localization.1–5 Here, we report evidence from two unrelated families that a novel missense variant in the adenosine triphosphate (ATP) synthase membrane subunit C locus 3 gene (ATP5MC3; formerly ATP5G3), may cause either condition. After initial linkage analysis in a large, multigenerational family (Family-1) with autosomal dominant inheritance, where affected members manifested with generalized dystonia and/or spastic paraplegia,6 genetic studies in this and a second family (Family-2) with a proband with generalized dystonia identified an identical de novo variant in ATP5MC3. We also report results of studies of function of the resulting protein. The structure of subunit c of the mitochondrial F0 portion of ATP synthase, in complex V of the electron transport chain, is highly conserved. A spontaneous mutation in the E. coli ortholog atpE at the same position uncouples ATP hydrolysis from proton translocation.7 Thus, we performed experiments to evaluate whether this human variant would similarly affect mitochondrial function and found supportive evidence in both patient cell lines and Drosophila.
Patients and Methods
Protocol Approval and Consents
Written informed consent, as approved by the Institutional Review Board of Cincinnati Children’s Hospital Medical Center, was obtained from all the participating members of Family-1. Additional dystonia and spastic paraplegia subjects were obtained under IRB approval by the University of Tennessee Health Science Center. For Family-2, written informed consent was obtained from the proband and parents under ethics review board-approved protocols from Technical University of Munich, Germany.
Whole Exome and Sanger Sequencing
Family-1 Genomic DNA was isolated from blood using Puregene reagents (Qiagen, Germantown, MD). Whole Exome Sequencing (WES) was performed in the proband and distant cousin with childhood-onset generalized dystonia, restricted to the previously mapped 2q24-31 locus.6 In the remaining family members, WES was performed by the Cincinnati Children’s DNA Sequencing and Genotyping Core. Exome preparation used NimbleGen EZ Exome (Roche, Indianapolis, IN). Sequences were read on an Illumina HiSeq2000 (San Diego, CA). Sanger sequencing was used to determine the ATP5MC3 mutation for segregation analysis. PCR was performed using primers (5’-TCAGTTTACAAAAATCCCCACA-3’) and (5’-CAGTTTTCGGAGTCACAGTAAGA-3’) and FastStart TAQ (Roche, Indianapolis, IN). PCR products were treated with ExoSAP-IT (Affymetrix, Santa Clara, CA) and sequenced using BigDye v1.1 chemistry on a 3730XL DNA Analyzer (Life Technologies, Grand Island, NY).
Family-2 parent-offspring trio WES was pursued using previously described methods.8, 9 Genomic DNA was isolated from whole blood and exonic sequences from DNA were enriched with the SureSelect Human All Exon 60Mb Kit (Agilent Technologies). Paired-end (100-bp) sequencing was done on a HiSeq4000 machine (Illumina) following manufacturer guidelines (sequencing parameters for the trio: generated raw data: 9.7Gb – 10.7Gb; mean coverage: 103x – 112x; >20x-coverage: 97.1% - 97.7%). For variant calling, annotation, and prioritization, we used an established, custom-developed bioinformatics pipeline (Helmholtz Center Munich and Technical University of Munich, Germany). Raw reads were mapped to human reference gene sequences based on genome build hg19/GRCh37 with the Burrows-Wheeler Aligner tool and variant calls were generated with the help of SAMtools, PINDEL, and ExomeDepth. Only calls that met high-confidence quality thresholds (read depth >10x, Phred quality score >30) were retained.
Bioinformatic Analysis
The Family-1 proband’s WES data was searched for rare (minor allele frequency < 0.005 in gnomAD v2.1.1 and ~20,000 in-house controls) protein-altering variants in previously described dystonia-related genes.8 Next, candidate disease-associated variants were prioritized under both dominant and recessive inheritance models. A single novel, heterozygous variant was identified: chr2:176,043,103C>G (GRCh37) resulting in a missense variant in ATP5MC3: NM_001689.5 c.318C>G (p.Asn106Lys) and was confirmed by bidirectional Sanger sequencing in the proband (Figure 1).
Figure 1. ATP5MC3 mutation and conservation analysis.
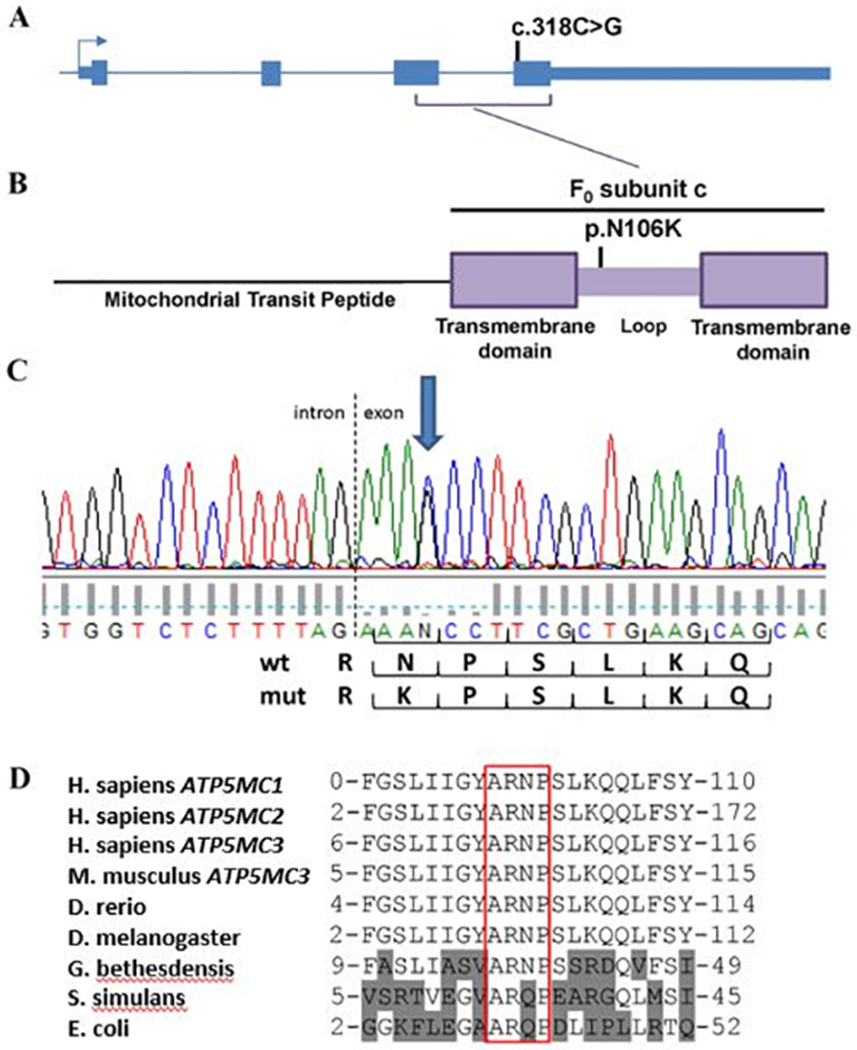
(A) c.318C>G mutation in exon 4 of ATP5MC3 resulting in (B) p.Asn106Lys substitution in the c subunit. (C) Electropherogram demonstrating heterozygous mutation near the start of exon 4. (D) ARNP motif conserved among c subunit genes APT5MC1–3, Drosophila, and bacteria including G. bethesdensis. Gln is substituted for Asn in S. simulans and E. coli.
To identify additional individuals with similar clinical phenotype and heterozygous variants in ATP5MC3, the Family-1 variant was submitted to GeneMatcher,10 where a match to Family-2 was identified.
Allele frequency was analyzed using the gnomAD v2.1.1 and v3.1.1 databases.11 Predictions of the pathogenicity were evaluated with SIFT/PROVEAN (http://sift.jcvi.org/), Mutation Taster (http://www.mutationtaster.org/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), and CADD (http://cadd.gs.washington.edu/score) software.12–16 Conservation alignments were performed using UCSC Genome Browser (http://genome.ucsc.edu/), Wormbase (wormbase.org), and Flybase (flybase.org).17–19
Screening for variants in additional individuals with undiagnosed dystonia and spastic paraplegia
Samples from patients with dystonia20 (n=362) and HSP (n=22) (see clinical data in Supplemental Table 1), who had been pre-screened and tested for prior known causes of dystonia and HSP, were evaluated by high resolution melting point (HRM) analysis to search for variants in the three ATP5MC genes (see Supplementary Table 2). The coding region of ATP5MC3 was Sanger sequenced in subjects with HSP.
HRM analyses were performed using the LightCycler® 480 Real-Time PCR system and High Resolution Master Mix from Roche Applied Science.20 Optimized HRM reactions were performed in 384-well plates using 20 ng of template DNA, 1X HRM Master Mix, 2.5 mM MgCl2 and 200 nM of each primer in a 10-μl reaction volume. Using LightCycler 480 Gene Scanning Software, melting curves and difference plots were analyzed. All samples were unambiguously assigned to genotypes by Gene Scanning software.
For sequencing, fourteen pairs of PCR primers were designed with Primer3 (frodo.wi.mit.edu) to encompass the coding regions of the three ATP5MC genes (Supplementary Table 3). Five μl of the PCR products were cleaned using ExoSAP-IT® (United States Biochemical, Cleveland, Ohio). Then, 1-2 μl of the purified PCR products were sequenced in the forward and reverse directions using the Applied Biosystems 3130XL Genetic Analyzer.
Drosophila production
ATPsynC cDNA (Annotation symbol: CG1746) was isolated from wild type embryonic RNA by RT-PCR using SuperScript III (Life Technologies) and primers (5’-GCCTGTCACCCAATATCTCC-3’) and (5’-TGCCAACATGTCAAGCAGTAA-3’). The cDNA was cloned into the TOPO-TA Cloning vector (Life Technologies) and modified by site-directed mutagenesis with QuikChange II (Agilent, Santa Clara, CA). Mutagenesis primers for the human-disease-equivalent p.N102K were (5’-CGGCTACGCCAGGAAGCCATCGCTGAAA-3’) and (5’-TTTCAGCGATGGCTTCCTGGCGTAGCCG-3’). For the E. coli equivalent mutation p.N102E, the primers were (5’-ATCGGCTACGCCAGGGAGCCATCGCTGAAACAG-3’) and (5’-CTGTTTCAGCGATGGCTCCCTGGCGTAGCCGAT-3’). The wild-type, p.N102K and p.N102E mutant constructs were then cloned into the pUAST-attB expression vector.21 Transgenic fly lines containing a single copy of each UAS construct, specifically integrated at the 51C locus on chromosome 2, were generated using phi3C integrase (performed by Rainbow Transgenic Flies, Inc, Camarillo, CA). Each UAS line was subsequently crossed to either the armadillo (arm)-Gal4, elav-Gal4, tub-Gal4, or 24B2-Gal4 driver lines available from the Bloomington Stock Center. All Drosophila lines were cultured and experiments performed at 25°C.
Human cell lines and culture conditions
Human fibroblast cells from the Family-1 proband and his father as well as from unrelated, age- and cell-passage matched, de-identified controls were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS) at 37 °C and with 5% CO2.
Drosophila mitochondria isolation
Mitochondria were isolated.22, 23 In brief, approximately 300 flies were immobilized by chilling on ice. 4 ml of ice-cold isolation medium (250 mM sucrose, 5 mM Tris-HCl, 2 mM EGTA, 1% (w/v) bovine serum albumin, pH 7.4 at 4°C) was added. The flies were gently crushed in cold isolation medium with a homogenizer and the homogenate centrifuged at 1000 g for 5 min at 4°C. The supernatant was recentrifuged at 10000 g for 10 min at 4°C and discarded and the mitochondrial pellet was saved.
Mitochondrial respiratory chain complex I-V activity assay, ATP measurement, and oxygen consumption rate
Methods were modified from previously published protocols.23 In brief, Complex activities were determined as follows: Complex I (NADH dehydrogenase) as the rotenone-sensitive NADH oxidation at 340 nm, using the coenzyme Q analogue 2,3-dimethyl-5-methyl 6-n-decyl-1,4- benzomethyluinone (DB) as an electron acceptor; Complex II (succinate dehydrogenase) by tracking the secondary reduction of 2, 6–dichlorophenolindophenol by ubiquinone-2 at 600 nm; Complex III (cytochrome bc1 complex) by measuring the reduction of cytochrome c at 550 nm with reduced decylubiquinone; Complex IV (cytochrome c oxidase) by monitoring the oxidation of reduced cytochrome c as a decrease of absorbance at 550 nm; Citrate synthase by the reduction of 5,5’- dithiobis-2-nitrobenzoic acid at 412 nm in the presence of acetyl- CoA and oxaloacetate. For analysis, Complex I–IV activities were normalized to citrate synthase activity. Complex V activity was measured by using the Mitochondrial Complex V Activity Assay kit (EMD Milipore, Billerica, MA) with modifications.24 The assay relies on linking the ATPase activity to NADH oxidation via the conversion of phosphoenolpyruvate to pyruvate by pyruvate kinase (PK) and then pyruvate to ATP production measurement.
The ATP level in cultured cells was determined using the ATP Bioluminescence Assay Kit CLS II (Roche, Indianapolis, IN). ATP was measured by luminescence signal and the ATP production was determined as luciferase activity22 and lactate by lactate dehydrogenase. The change in absorbance was recorded at 340 nm.
The oxygen consumption rate (OCR) measurements were performed by using a Seahorse Bioscience XF-24 instrument (Seahorse Biosciences, North Billerica, MA, USA). Cells were seeded in XF24-well micro plates in growth medium, which was replaced the next day with 675 μl of assay medium.23
Testing of effects of ATP5MC3 mutations on Drosophila mobility
Drosophila mobility was evaluated using geotaxis in a ten chamber countercurrent device.25 Flies were phenotypically sorted with humidified carbon dioxide anesthesia and then returned to a 25°C incubator for a 24 hour recovery period prior to testing. Thirty to fifty flies were introduced into the first lower chamber, tapped to the bottom, and observed for 15 seconds as they climbed the 8 cm distance into the top chamber. At each 15 second interval, the upper chambers were shifted along a sliding mechanism, the flies tapped to the bottom, and the upper chambers returned to their initial position (Supplementary video). Thus, fast climbers enter the upper chamber and are progressively transferred into adjoining chambers, whereas slower flies remain in the same tube. Results are presented as “tube distance,” which represents the average lateral distance traveled, but can be understood as the average number of cycles that a population of flies will travel to the upper chamber.
Results
Clinical information for Family-1
Dystonic and spastic paraplegic phenotypes of the affected individuals were previously reported.6 In brief, the proband had unilateral upper limb onset at age 2 years that generalized to trunk and all limbs, resulting in loss of ambulation. Bilateral pallidal deep brain stimulation, implanted at age 9 years, restored ambulation and limb use. At age 25 years, his response remains excellent. The proband’s father and grandfather manifest the HSP phenotype with onset of progressive leg weakness and spasticity after age 20 years. The autosomal dominant pedigree included 40 individuals, of which 18 were affected with either childhood-onset, progressive generalized dystonia or adult-onset, gradually progressive spastic paraplegia. Less severe, later-onset, focal limb and neck dystonia and spasmodic dysphonia also occurred, possibly related to trauma or overuse. Brain MRIs and peripheral nerve physiology, when performed, were normal. Metabolic studies, including whole blood lactate and pyruvate, serum amino acids, urine organic acids, and acylcarnitine profiles from blood and urine were normal in both the proband and his father.
Clinical information for Family-2
A second family with the c.318C>G (p.Asn106Lys) variant was independently identified (Family-2). The proband of this family was a right-handed male, born to healthy unrelated parents of European descent with no family history of dystonia or related movement disorders, who first presented to the Department of pediatric neurology in Freiburg (Germany) at the age of 11 years with a 4-year history of gradually worsening upper limb dystonia. The proband in this family was diagnosed with a childhood-onset dystonic syndrome and enrolled in a research project focusing on the molecular bases of dystonia (Helmholtz Center Munich and Technical University of Munich, Germany). The proband`s progressive condition prompted a comprehensive workup. However, all laboratory studies of blood, urine, and cerebrospinal fluid and testing for dystonia-causing mutations via gene panel analysis were unrevealing. Brain MRI revealed nonspecific mild frontoparietal white-matter changes bilaterally without other associated abnormalities. However, parent-offspring trio WES revealed a NM_001689.4:c.318C>G (p.Asn106Lys) variant as a heterozygous, de novo mutation in the proband (Supplementary Figure 1).
Candidate gene analyses
Whole exome analysis on the proband and distant cousin with childhood onset dystonia, restricted to novel shared coding variants within the linkage region, identified a single missense variant, (GRCh37/hg19, NC_000002.11:g.176043127G>C, NM_001689.4:c.318C>G, NP_001680.1:p.(Asn106Lys), within ATP5MC3. Consistent with the pedigree phenotypes, Sanger sequencing showed only affected individuals carrying the ATP5MC3 variant.
ATP5MC3 encodes for the transmembrane-spanning subunit c of the F0 portion of mitochondrial ATP synthase. The substituted amino acid exists in a highly conserved loop structure that directly interacts with the F1 subunit (Fig 1).26 This interaction produces the coupled rotation needed to transduce the potential energy of the proton gradient into the phosphorylation of ADP.27 This loop structure is conserved in all vertebrates examined, as well as C. elegans (Y82E9BR.3) and Drosophila melanogaster (CG1746). The p.Asn106Lys variant has not been documented in gnomAD v2.1.1 or v3.1.1.11 Computational analyses predict the proband’s variant to be damaging by all metrics evaluated, including PolyPhen2 HDIV-score (1.0), MutationTaster score (1), REVEL rankscore (0.75), PROVEAN score (score -5.5), SIFT score (score 0.0), and CADD PHRED score (25.4, GRCh37-v1.6; 24.7, GRCH38-v1.6). Two other extremely rare variants altering Asn106 have been identified, in one individual each, in gnomAD v2.1.1 (p.Asn106Ser) and in v3.1.1 (p.Asn106Tyr). While these are also predicted to be damaging, it is unknown whether these variants were associated with neurological symptoms. Globally, ATP5MC3 is not highly constrained to missense variation (67% of the expected number missense variants were observed in gnomAD v2.1.1), but such constraint would not be expected for a condition that does not limit reproductive fitness.
ATP5MC1 and ATP5MC2 also encode for subunit c. These proteins differ in their N-terminal mitochondrial import sequences while their C-terminal cleavage products, which give rise to the mature subunit c, remain identical.26 To identify additional subunit c variants in patients with dystonia or spastic paraplegia, 384 patients were screened for variants in ATP5MC1, ATP5MC2, and ATP5MC3. No deleterious variants were identified in the cohort of patients with dystonia or spastic paraplegia (Supplementary Table 1). Identified sequence variants are included in Supplementary Table 2.
Mitochondrial studies
In fibroblasts from skin biopsies from the Family-1 proband (dystonia) and his affected father (HSP), activities of electron transport chain complexes I through IV were equivalent to controls. However, Complex V activity was reduced by 20% (p < 0.01) (Figure 2). Similarly, cellular ATP was reduced 20% to 32% and oxygen consumption 21% to 25% (Figure 2). As loss of one of six genomic copies of ATP5MC1/2/3 would result in an expected reduction of approximately 17% of complex V activity, these results support the predicted pathogenicity of the p.Asn106Lys mutation. In E. coli, subunit c of ATP synthase is encoded by atpE. A spontaneous mutation of the equivalent amino acid p.Gln42Glu leads to uncoupling of ATP hydrolysis from proton translocation.7 Uncoupling of oxidative phosphorylation, however, was not supported as a mechanism for the human mutation, as both oxygen consumption and ATP were proportionally reduced.
Figure 2. Decreased complex V function in ATP5MC3-associated disease.
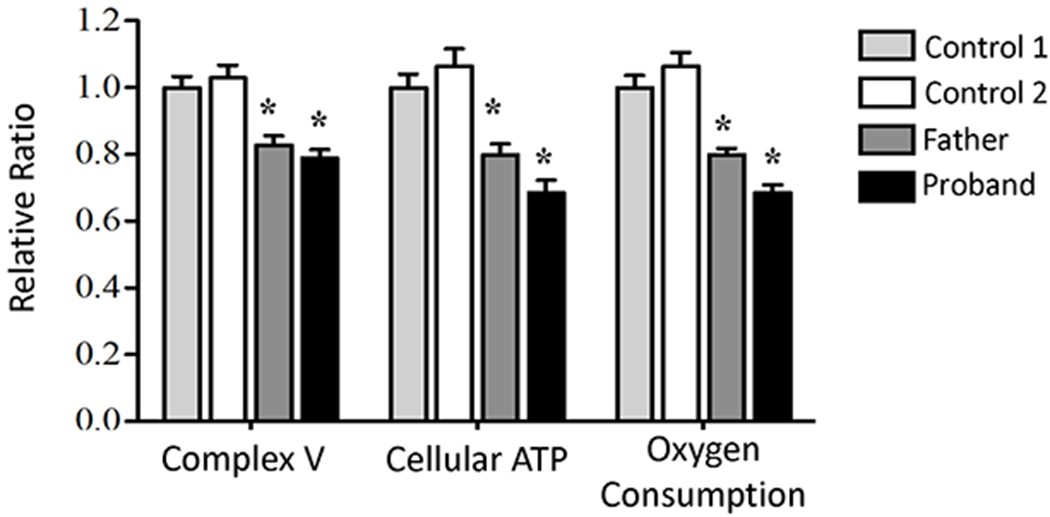
A significant decrease was observed in relative ATP synthase activity in fibroblast cell lines derived from proband and affected father compared to two unrelated control fibroblast cell lines, with an associated decrease in cellular ATP and oxygen consumption in patient fibroblast cells lines. (* p-value <0.01).
Drosophila mutation constructs and geotaxis
Drosophila have one homologous copy of ATP5MC3 (CG1746). Tissue-specific expression of wild type and mutant ATP5MC3 proteins was performed using the GAL4-UAS transgenic system in which the UAS enhancer sequence activates gene expression in the presence of GAL4.28 Stable transgenic flies were produced with wild type and mutated forms of CG1746 (p.N102K and p.N102E) under control of UAS. Multiple GAL4 drivers were tested. The muscle-specific 24B2 and the neuronal-specific elav GAL4 drivers produced no discernible phenotypic effects. The constitutive tubulin-Gal4 driver was non-viable in all expression lines, including wild-type, precluding further characterization (data not shown). The armadillo (arm) driver (homologous to human beta-catenin) provided a milder constitutive driver with demonstrable phenotypic effect.
The UAS-response elements were inserted on chromosome 2 and arm-GAL4 on chromosome 3, allowing for breeding strategies to produce homozygous alleles on both chromosomes. Double homozygous strains of the arm-GAL4 and UAS-p.N102K were non-viable. As such, flies bearing a single copy of arm-GAL4 and homozygous copies of the UAS-response elements were used.
At less than one week of age, the human disease mutant subunit c (p.N102K) and the equivalent bacterial mutation7 (p.N102E) demonstrated reduced mobility relative to wild type flies and the arm-GAL4; UAS-WT construct (Fig 3A; Supplementary Video). In serial experiments, Age and Genotype both reduced geotaxis (F4, 1873 = 292.2; p<0.0001). Genotype had a larger effect than Age; the Age*Genotype interaction was non-significant (Fig 3B). Both mutants were slower than wildtype (p <0.0001). The human p.N102K mutant was slowest.
Figure 3. Drosophila geotaxis assay.
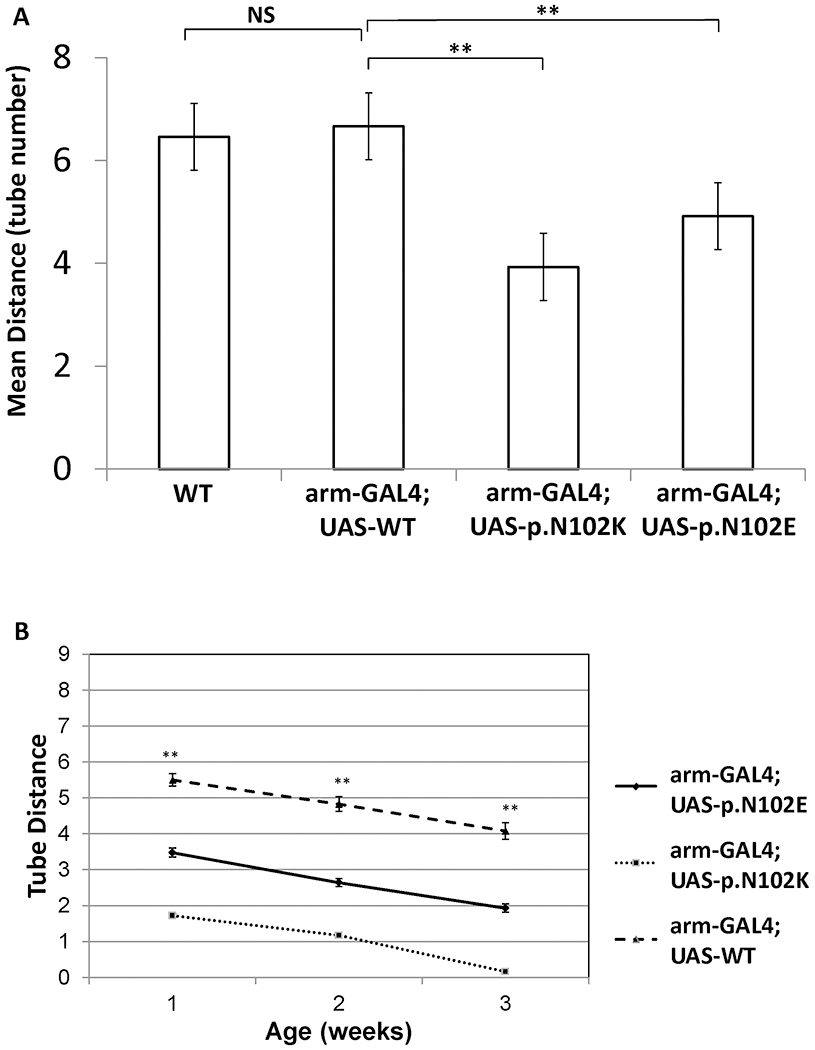
(A) Mean distance traveled in geotaxis assay for transgenic flies with arm-GAL4 driver with or without UAS constructs (see also supplementary video). Wild type flies compared to UAS-WT overexpression flies showed no significant difference. Flies carrying p.N102K and p.N102E expression constructs traveled significantly shorter distances. (B) Aging flies showed decreasing activity with age but demonstrated consistent differences between transgenic lines. (** p-value <0.001).
In Drosophila mitochondria, concordant with the results for geotaxis, the Complex V activity was reduced by 53% for the human p.N102K mutant (p=0.0008) and by 37% for the bacterial p.N102E mutant (p=0.003) (Fig 4). In negative control lines, Complex V activity was similar to wild type for all UAS-response elements without expression drivers, further supporting that the ATP5MC3 variant (c.318C>G, p. Asn106Lys) is pathogenic.
Figure 4. Drosophila complex V activity in transgenic flies.
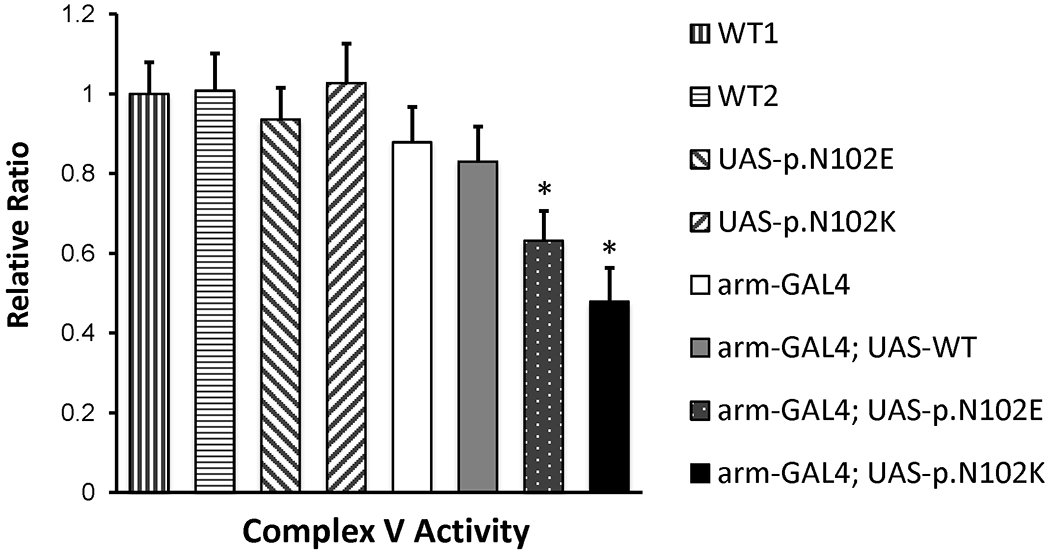
Relative ATP synthase activity among control and UAS-GAL4 transgenic flies. Significant reductions were noted in flies containing both arm-GAL4 driver and UAS constructs with either p.N102E or p.N102K mutations, but not with wild type. WT1 is a yellow mutant, which retains normal neuromuscular function. WT2 contains the CyO (curly wing) balancer used to create UAS-GAL4 transgenic lines. (* p-value <0.01; ** p-value <0.001).
Discussion
In a large, single-family pedigree manifesting an unusual combination of autosomal dominant dystonia and spastic paraplegia, we report the presence of a novel variant in ATP5MC3, a nuclear gene which encodes the c subunit of mitochondrial Complex V. Dystonia in this pedigree can generalize if manifest in childhood, shows incomplete penetrance, and responds to pallidal deep brain stimulation. The spastic paraplegia presents in adulthood, progresses gradually, and is not accompanied by other neurological problems. The pathogenicity of this variant for dystonia has been confirmed by the identification of the same variant in a patient with upper limb dystonia from a second, independent family. ATP5MC3 remains the primary candidate for the spastic paraplegia in the studied family and should be considered a target for further study, with evidence for causality based on 2015 American College of Medical Genetics and Genomics/Association for Molecular Pathology Standards and Guidelines29 including strong functional studies, low frequency in the general population, supportive in silico predictions, and cosegregation.30
To date, variants in ATP5MC3 have not been reported in other human diseases, nor could we identify a similar mutation in any additional patients manifesting dystonia or spastic paraplegia. To further address the pathogenicity of this variant, we undertook additional experiments to elucidate function and clarify possible pathophysiological mechanisms.
ATP5MC3 encodes a component of ATP synthase (electron transport chain complex V), which is critical to oxidative phosphorylation, the aerobic process by which energy substrates are converted to ATP.26 The electron transport chain uses energy substrates to form an electrochemical gradient of protons. ATP synthase then uses the proton gradient as an energy source to drive conversion of ADP to ATP. To do this, the protons are translocated across the inner mitochondrial membrane by the F0 subunit of complex V. As the protons traverse, they induce a structural change in F0 causing its rotor component to engage and rotate the F1 subunit. This rotation brings ADP and inorganic phosphate into alignment, allowing phosphorylation.31 ATP5MC3 encodes the monomer, subunit c, which aggregates to form the F0 rotor. The variant in these two families substitutes asparagine for lysine at position 106, in a loop of ATP5MC3 with complete homology from human to drosophila. This conserved loop acts as the point of contact between F1 and F0.32 As such, we hypothesized that the p.Asn106Lys variant would impair oxidative phosphorylation by disrupting this point of contact.
Understanding pathophysiology of this variant should account for the fact that two paralogs, ATP5MC1 and ATP5MC2, also encode subunit c. These functionally identical proteins have divergent mitochondrial import sequences which are cleaved following import.33 Assuming equal expression of the three alleles, haploinsufficiency would be unlikely to account for the severe phenotypes, as 83% of the active protein would still remain. Thus, we hypothesized that the presence of the mutant allele interfered with the F1-F0 interaction which could be tested through overexpression.
To this end we conducted several experiments, all of which support the pathogenicity of this ATP5MC3 mutation. First, in vitro functional studies of fibroblasts from the proband of Family-1 and his father demonstrated a reduction of complex V activity. Second, overexpression of the mutant form of the homolog in Drosophila resulted in statistically significant impairment of mobility (geotaxis), whereas overexpression of wildtype protein did not. Third, as a naturally occurring mutation, p.N102E, in the homologous E. coli gene atpE reduces Complex V function and ATP production (while preserving proton translocation),7 we studied this mutation and found that both Drosophila geotaxis and complex V activity were significantly reduced, although to a lesser extent than with the human mutation. Finally, the human mutation reduced complex V activity in Drosophila with a severity that matched that observed motor slowing in the geotaxis assays. Taken together, these experiments strongly support the pathogenicity of this ATP5MC3 variant.
Further study may clarify mechanisms of phenotypic heterogeneity. Ten to fourteen copies of subunit c create the transmembrane spanning domain of F0. The relative composition of normal versus mutant subunit c in the overall structure will likely influence the function of complex V. In Drosophila, different GAL4 drivers and their relative dosage produced different degrees of severity. Thus, we suspect that relative expression of the mutated gene may mediate phenotypic expression. The specific regulatory control of ATP5MC genes remains unstudied, but expression assays suggest that the expression levels of ATP5MC1, 2, and 3 respond independently when exposed to the same stressor.34–40 Differential expression patterns for ATP5MC genes within the nervous system (e.g. striatum versus spinal cord) or resulting from environmental factors such as oxidative stress,39–42 might increase the proportion of mutant ATP5MC3, influencing the phenotype.
Although this pedigree has unusual features, other dystonias and spastic paraplegias may involve similar pathophysiological mechanisms.3 For example, variants in MTATP6 cause mitochondrially-inherited neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP) syndrome,43 infantile bilateral striatal necrosis,44 and Leigh Syndrome.45 Two extremely rare and severe autosomal recessive diseases, resulting from mutations in transmembrane protein TMEM7046 and ATPAF2,47 involve defective F1 complex assembly. Other mitochondrial diseases can also present with secondary dystonia.48 Spastic paraplegias involving mitochondrial pathology can be pure or complex.49 These include a late onset HSP-like disorder linked to ATP synthase 650 as well as a variable onset, autosomal dominant variant of neurodegeneration with iron accumulation type-4, NBIA/HSP-C19orf12, due to deletion the C19ORF12.51, 52
In conclusion, in a large pedigree with an unusual autosomal dominant form of dystonia and spastic paraplegia, we identified a cosegregating p. Asn106Lys variant of ATP5MC3. The association with dystonia has been confirmed in a second family, supporting pathogenicity for the dystonia phenotype. Functional experimental studies show this mutation reduces complex V activity and, in a Drosophila model, impairs mobility. Further studies are needed to clarify the mechanisms whereby disorders involving energy metabolism may induce neurological disease with varying phenotypes.
Supplementary Material
Video legend. Geotaxis assay. Drosophila shown at weeks 1 and 3. Wild type and human variants, as labelled. Beginning and end of each of 4 assays shown due to file-size limitations.
Acknowledgements
BG is funded by a grant (GM079428) from the National Institutes of Health (NIH). MSL was supported by NIH grants R01 NS069936 and R01 NS082296, the Dystonia Medical Research Foundation and Benign Essential Blepharospasm Research Foundation. TH was supported by the Cincinnati Children’s Research Foundation. DLG is funded by the Bray Family Dystonia Research Fund and the Cincinnati Children’s Hospital Medical Center Movement Disorders Research fund. This study was also funded in part by in-house institutional funding from Technische Universität München, Munich, Germany, Helmholtz Zentrum München, Munich, Germany. MZ and JW receive research support from the German Research Foundation (DFG; ZE 1213/2-1; WI 1820/14-1).
This work was previously presented in part at the 65th Annual Meeting of the American Academy of Neurology, San Diego, CA, March 21, 2013; at the 17th International conference of Parkinson’s disease and Movement Disorders, Sydney, Australia June 19, 2013; and at the 63rd Annual Meeting of the American Society of Human Genetics, Boston, MA, October 22, 2013.
Footnotes
Financial Disclosures
Dr. Neilson
Dr. Zech: No disclosures.
Dr. Hufnagel: No disclosures.
Dr. Slone: No disclosures.
Dr. Wang: No disclosures.
Ms. Homan: No disclosures
Ms. Gutzwiler: No disclosures.
Dr. E. Leslie: No disclosures.
Dr. N Leslie: No disclosures.
Dr. Xiao: No disclosures.
Dr. Hedera: Dr. Hedera has served on the scientific advisory board for Alexion. He has received funding for travel/speaker honoraria from Ipsen and for serving on the speakers’ bureaus for Ipsen and Abbvie. He serves as an associated editor for Neurology.
Dr. LeDoux has been a consultant for USWorldMeds; speaker for Adamas Pharmaceuticals, Acadia Pharmaceuticals, Teva Pharmaceutical Industries, Amneal, USWorldMeds, Supernus, Kyowa Kirin, and Acorda Therapeutics; and receives publishing royalties from Elsevier (Animal Models of Movement Disorders, and Movement Disorders: Genetics and Models) and TheBookPatch (Parkinson’s Disease Poetry). Dr. LeDoux’s research has been funded by the Michael J. Fox Foundation, National Institutes of Health, Axovant Sciences, Wave Life Sciences, Teva Pharmaceutical Industries, Pharma Two B, Revance, Cerevel, Aeon, NeuroDerm, Dystonia Medical Research Foundation, and Benign Essential Tremor Research Foundation.
Dr. Gebelein: No disclosures.
Dr. Wilbert: No disclosures.
Dr. Eckenweiler: No disclosures.
Dr. Winkelmann: No disclosures.
Dr. Gilbert has received honoraria and/or travel support from the Tourette Association of America/ Centers for Disease Control and Prevention, the Child Neurology Society, and the American Academy of Neurology. He has received compensation for expert testimony for the U.S. National Vaccine Injury Compensation Program, through the Department of Health and Human Services. He has received payment for medical expert opinions through Advanced Medical/Teladoc. He has served as a consultant for Applied Therapeutics and Eumentics Therapeutics. He has received research support from the NIH (NIMH) and the DOD. He has received salary compensation through Cincinnati Children’s for work as a clinical trial site investigator from Emalex (clinical trial, Tourette Syndrome) and EryDel (clinical trial, Ataxia Telangiectasia). He has received book/publication royalties from Elsevier, Wolters Kluwer, and the Massachusetts Medical Society.
Dr. Huang: No disclosures
References
- 1.Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Movement disorders : official journal of the Movement Disorder Society 2013;28(7):863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Draganski B, Bhatia KP. Brain structure in movement disorders: a neuroimaging perspective. Current Opinion in Neurology 2010;23(4):413–419. [DOI] [PubMed] [Google Scholar]
- 3.Blackstone C Converging cellular themes for the hereditary spastic paraplegias. Curr Opin Neurobiol 2018;51:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parodi L, Fenu S, Stevanin G, Durr A. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev Neurol (Paris) 2017;173(5):352–360. [DOI] [PubMed] [Google Scholar]
- 5.Klebe S, Stevanin G, Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting. Rev Neurol (Paris) 2015;171(6–7):505–530. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert DL, Leslie EJ, Keddache M, Leslie ND. A novel hereditary spastic paraplegia with dystonia linked to chromosome 2q24–2q31. Mov Disord 2009;24(3):364–370. [DOI] [PubMed] [Google Scholar]
- 7.Mosher ME, White LK, Hermolin J, Fillingame RH. H+-ATPase of Escherichia coli. An uncE mutation impairing coupling between F1 and Fo but not Fo-mediated H+ translocation. The Journal of biological chemistry 1985;260(8):4807–4814. [PubMed] [Google Scholar]
- 8.Zech M, Boesch S, Jochim A, et al. Clinical exome sequencing in early-onset generalized dystonia and large-scale resequencing follow-up. Mov Disord 2017;32(4):549–559. [DOI] [PubMed] [Google Scholar]
- 9.Zech M, Boesch S, Maier EM, et al. Haploinsufficiency of KMT2B, Encoding the Lysine-Specific Histone Methyltransferase 2B, Results in Early-Onset Generalized Dystonia. American journal of human genetics 2016;99(6):1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Human mutation 2015;36(10):928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581(7809):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nature methods 2010;7(4):248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods 2010;7(8):575–576. [DOI] [PubMed] [Google Scholar]
- 14.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PloS one 2012;7(10):e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols 2009;4(7):1073–1081. [DOI] [PubMed] [Google Scholar]
- 16.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics 2014;46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marygold SJ, Leyland PC, Seal RL, et al. FlyBase: improvements to the bibliography. Nucleic acids research 2013;41(Database issue):D751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome research 2002;12(6):996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yook K, Harris TW, Bieri T, et al. WormBase 2012: more genomes, more data, new website. Nucleic acids research 2012;40(Database issue):D735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao J, Zhao Y, Bastian RW, et al. Novel THAP1 sequence variants in primary dystonia. Neurology 2010;74(3):229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proceedings of the National Academy of Sciences of the United States of America 2007;104(9):3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manfredi G, Yang L, Gajewski CD, Mattiazzi M. Measurements of ATP in mammalian cells. Methods 2002;26(4):317–326. [DOI] [PubMed] [Google Scholar]
- 23.Mullen PJ, Zahno A, Lindinger P, et al. Susceptibility to simvastatin-induced toxicity is partly determined by mitochondrial respiration and phosphorylation state of Akt. Biochimica et biophysica acta 2011;1813(12):2079–2087. [DOI] [PubMed] [Google Scholar]
- 24.Kwong JQ, Davis J, Baines CP, et al. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell death and differentiation 2014;21(8):1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryan EL, DuBoff B, Feany MB, Fridovich-Keil JL. Mediators of a long-term movement abnormality in a Drosophila melanogaster model of classic galactosemia. Disease models & mechanisms 2012;5(6):796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan WL, Lerner TJ, Haines JL, Gusella JF. Sequence analysis and mapping of a novel human mitochondrial ATP synthase subunit 9 cDNA (ATP5G3). Genomics 1994;24(2):375–377. [DOI] [PubMed] [Google Scholar]
- 27.Sebald W, Hoppe J. On the structure and genetics of the proteolipid subunit of the ATP synthase complex. Curr Topics Bioenerget 1981;12:1–64. [Google Scholar]
- 28.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993;118(2):401–415. [DOI] [PubMed] [Google Scholar]
- 29.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarvik GP, Browning BL. Consideration of Cosegregation in the Pathogenicity Classification of Genomic Variants. Am J Hum Genet 2016;98(6):1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Ballmoos C, Wiedenmann A, Dimroth P. Essentials for ATP synthesis by F1F0 ATP synthases. Annual review of biochemistry 2009;78:649–672. [DOI] [PubMed] [Google Scholar]
- 32.Hermolin J, Dmitriev OY, Zhang Y, Fillingame RH. Defining the domain of binding of F1 subunit epsilon with the polar loop of F0 subunit c in the Escherichia coli ATP synthase. The Journal of biological chemistry 1999;274(24):17011–17016. [DOI] [PubMed] [Google Scholar]
- 33.Vives-Bauza C, Magrane J, Andreu AL, Manfredi G. Novel role of ATPase subunit C targeting peptides beyond mitochondrial protein import. Molecular biology of the cell 2010;21(1):131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anitha A, Nakamura K, Thanseem I, et al. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain pathology 2013;23(3):294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu TJ, Peters DG. Serial analysis of the vascular endothelial transcriptome under static and shear stress conditions. Physiological genomics 2008;34(2):185–192. [DOI] [PubMed] [Google Scholar]
- 36.Huang Y, Wang L, Bennett B, et al. Potential role of Atp5g3 in epigenetic regulation of alcohol preference or obesity from a mouse genomic perspective. Genetics and molecular research : GMR 2013;12(3):3662–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishihata A, Katano Y. Investigation of differentially expressed genes in the ventricular myocardium of senescent rats. Annals of the New York Academy of Sciences 2006;1067:142–151. [DOI] [PubMed] [Google Scholar]
- 38.Kim JW, Kwon OY, Kim MH. Differentially expressed genes and morphological changes during lengthened immobilization in rat soleus muscle. Differentiation; research in biological diversity 2007;75(2):147–157. [DOI] [PubMed] [Google Scholar]
- 39.Merscher S, Funke B, Epstein JA, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001;104(4):619–629. [DOI] [PubMed] [Google Scholar]
- 40.Parikh H, Nilsson E, Ling C, et al. Molecular correlates for maximal oxygen uptake and type 1 fibers. American journal of physiology Endocrinology and metabolism 2008;294(6):E1152–1159. [DOI] [PubMed] [Google Scholar]
- 41.Ramlawi B, Otu H, Mieno S, et al. Oxidative stress and atrial fibrillation after cardiac surgery: a case-control study. The Annals of thoracic surgery 2007;84(4):1166–1172; discussion 1172–1163. [DOI] [PubMed] [Google Scholar]
- 42.Hong Y, Piao F, Zhao Y, Li S, Wang Y, Liu P. Subchronic exposure to arsenic decreased Sdha expression in the brain of mice. Neurotoxicology 2009;30(4):538–543. [DOI] [PubMed] [Google Scholar]
- 43.Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. American journal of human genetics 1990;46(3):428–433. [PMC free article] [PubMed] [Google Scholar]
- 44.De Meirleir L, Seneca S, Lissens W, Schoentjes E, Desprechins B. Bilateral striatal necrosis with a novel point mutation in the mitochondrial ATPase 6 gene. Pediatric neurology 1995;13(3):242–246. [DOI] [PubMed] [Google Scholar]
- 45.Castagna AE, Addis J, McInnes RR, et al. Late onset Leigh syndrome and ataxia due to a T to C mutation at bp 9,185 of mitochondrial DNA. American journal of medical genetics Part A 2007;143A(8):808–816. [DOI] [PubMed] [Google Scholar]
- 46.Cizkova A, Stranecky V, Mayr JA, et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nature genetics 2008;40(11):1288–1290. [DOI] [PubMed] [Google Scholar]
- 47.De Meirleir L, Seneca S, Lissens W, et al. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. Journal of medical genetics 2004;41(2):120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thompson VB, Jinnah HA, Hess EJ. Convergent mechanisms in etiologically-diverse dystonias. Expert opinion on therapeutic targets 2011;15(12):1387–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noreau A, Dion PA, Rouleau GA. Molecular aspects of hereditary spastic paraplegia. Experimental cell research 2014;325(1):18–26. [DOI] [PubMed] [Google Scholar]
- 50.Verny C, Guegen N, Desquiret V, et al. Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion 2011;11(1):70–75. [DOI] [PubMed] [Google Scholar]
- 51.Hartig MB, Iuso A, Haack T, et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. American journal of human genetics 2011;89(4):543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta neuropathologica 2013;126(3):307–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video legend. Geotaxis assay. Drosophila shown at weeks 1 and 3. Wild type and human variants, as labelled. Beginning and end of each of 4 assays shown due to file-size limitations.